
|
|
Stochastic dynamics of macromolecular assemblies
|
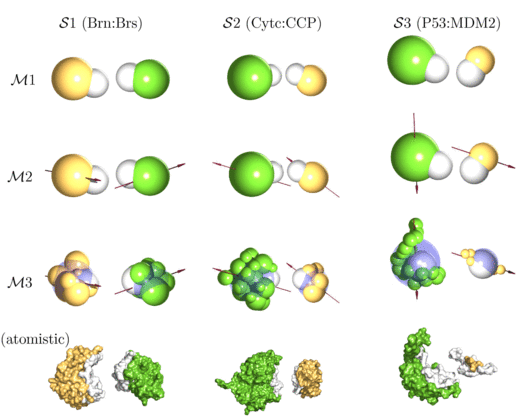
| Multi-scale modelling approach to protein-protein interactions: this figure shows different coarse-graining strategies (M1-M3) for different bimolecular systems of interest (S1-S3). The bottom row shows the full atomistic structures taken from the protein database. |
Proteins are the workhorses of the cell and therefore a large body of research has been focused on the physical principles underlying their function. The protein folding problem has been solved to a certain extend (at least on a practical level) and the new frontier is now to understand how proteins interact with each other in the cell. Proteomics provides us with a more or less complete list of the involved players and their possible interactions, but we are still missing the grand picture of how so many different and diverse components act together to yield robust and reliable results. The key to this problem is certainly temporal and spatial coordination, therefore it is high time to revisit generic models for the way proteins interact in time and space, and to augment them with our newly gained high-throughput knowledge.
In our work, we use a Langevin equation approach to arrive at computational tractable models for the dynamics of large protein complexes. There are many large complexes in the cell which are highly dynamic, for example adhesion clusters, the endocytotic complex, the mitotic spindle, and nuclear pore complexes. In order to arrive at models for such large systems, one cannot work at atomic resolution. On the other hand, information resulting from the molecular level has to be kept to a certain extend to ensure specificity of the model. To solve this problem, we use a rather generic, but quite flexible model of reaction patches placed on model proteins. Protein shape and interactions are derived from known structures and molecular dynamics simulations, respectively. Special care is taken to define rules for encounter and complex formation. Once parameterized in this way, our models then can be used to study the statistics in space and time of larger clusters.
As a first application, we have studied bimolecular association of three model systems with distinctly different properties: (S1) barnase:barstar, (S2) cytochrome c:cytochrome c peroxidase and (S3) p53:MDM2. In each case, proteins are modeled either as (M1) spherical particles, as (M2) dipolar spheres or as (M3) collection of several small beads with one dipole. The nine different model systems are shown in the figure above together with the atomistic structures taken from the protein data bank. Our study shows how generic principles of protein-protein association are modulated by molecular features of the systems under consideration and paves the way to deal with larger systems in the future (Journal of Chemical Physics 2008).
A Langevin equation approach for particles with reaction patches has also been used to study the role of shape for protein-protein encounter. We found that for realistic parameter values, the crossover from anisotropic to isotropic diffusion is much faster than typical encounter times (Physical Review E 2010).
Published work:
- J. Schluttig, D. Alamanova, V. Helms, and U. S. Schwarz. Dynamics of protein-protein encounter: A Langevin equation approach with reaction patches. J. Chem. Phys., 129:155106, 2008. (abstract, qbio arXiv 0809.2871, DOI:10.1063/1.2996082, PDF)
- J. Schluttig, C C. Korn, and U. S. Schwarz. Role of anisotropy for protein-protein encounter Phys. Rev. E, 81: 030902(R), 2010. (abstract, qbio arXiv 1002.4485, doi:10.1103/PhysRevE.81.030902, PDF)
Last modified Di 4. Okt 17:45:17 CEST 2011
by USS.
Back to home page Ulrich Schwarz.